

|
|

|
Промышленный лизинг
Методички
СЦу Ciz 1  Рис. 2.13. Кривые, характеризующие свойства эластомеров при гармонических колебаниях: а - петля гистерезиса; б - угол сдвига фаз; в - составляющие динамического модуля состоянии полимера а и е совпадают по фазе. Угол ф максимален в переходной области, в которой период Т* сравним с временем релаксации т. В области 9 > &с при каждом цикле деформации необратимо затрачивается работа, характеризуемая площадью петли гисте-реза (рис. 2.13, а); удельные потери энергии за цикл W= dz яаово sin ф. Потери в единицу времени, или плотность мощности потерь, Вт/м: N<p = WIT* = 0,5соаоео sin ф. (2.5) Комплексный модуль Е* состоит из вещественной части £, характеризующей упругость материала и мнимой части Е", характеризующей внутреннее трение: £оо + Eh Е* = Е + Е"; Е = 1 -I- юЧ „ (£у-н£)сот " l-1-coV • Теплофизические свойства. Соотно-щение между удельным объемом v (или плотностью р), давлением р и температурой 9 устанавливают уравнения состояния (1.1) - (1.4), в которые входят функциональные температурные коэффициенты объемного и линейного рас-щирения (X и щ, модуль объемного сжатия и или сжимаемость Р = 1/х. Значения а, X, теплоемкости с, теплопроводности X, и других теплофизических параметров необходимо знать во всем диапазоне р и 9 работы материалов уплотнений. Обычно известны только эксперимен- тальные данные об этих параметрах при нормальных условиях (ро, 9о), причем для ограниченного числа материалов. При описании принципиальных зависимостей а, X,... от р и 9 для полимеров в вязкоте-кучем, высокоэластическом и аморфном состояниях в первом приближении можно использовать модели физических свойств жидкости (см. подразд. 1.2). Все теплофизические свойства связаны с молекулярной структурой и свободным объемом Vf = F (р, 9) вещества. С повы-щением р V} уменьщается, поэтому следует ожидать увеличения х и уменьще-ния а. Обратное влияние оказывает температура. Большое гидростатическое давление р влияет на температуры фазовых переходов - стеклования 9„ плавления 9ш1, кристаллизации 9кр. С увеличением давления (рис. 2.14) температура 9с повышается, причем для эластомеров d9c/dp =0,17°С/МПа; для НК - 0Д4; для полиизобутилена - 0,25; для аморфных полимеров: 0,32 (ПС); 0,2-0,29 (ПММА); 0,11-0,13 (ПВХ) [3]. Изменение структуры материала при переходе через точки 9с 9ш„ 9кр вызы- 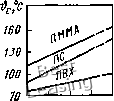 0 50 100 р,МПа Рис. 2.14. Зависимость температуры стеклования 9о полимеров от давления -120 - 80 -40 О 40 0,°С Рис. 2.15. Температурная зависимость коэффициента объемного расширения каучука НК вает ступенчатое изменение кривых теплофизических параметров. Температурный коэффициент объемного расширения а, °С-, определяют [3] из уравнения Тейта (1.4): где ро, р - плотности в исходном и рабочем режимах; В = Во-* « Во - с9 -функциональный коэффициент. В аморфном состоянии для ПС Во = 334 МПа и с =1,1 МПа/°С; для ПММА - 370 и 1,47; для ПВХ - 375 и 0,9; для ПЭВП -476 и 2,3; для ПЭНП - 323 и 2,26 [3]. В области перехода (9) к высокоэластическому состоянию Во резко уменьщается до 150 - 200 МПа и соответственно ступенчато возрастает а (рис. 2.15). Температурный коэффициент линейного расширения = а/3. Приближенную оценку коэффициента а при р и 9 по известному для начальных условий ао (ро = 0,1 МПа; 9о = 20...50 °С) выполняют по формуле а = (ХоХо/к, (2.7) где Ко, X - модули объемного сжатия соответственно при ро, 9о и р, 9. Уравнение (2.7) можно применять в пределах диапазона А9 между точками фазовых переходов. При переходе полимера из высокоэластического состояния в аморфное или частично-кристаллическое ос резко уменьшается (см. рис. 2.15). Кривые зависимостей а (9) и а(р) для некоторых полимеров имеют ступенчатые переходы, отражающие частичные структурные изменения. В
Рис. 2.16. Зависимость относительного объема ДК/К от давления р при й = 20... 25 °С (по опытам Бриджмена): / - эластомеры; 2 - фторопласт особенности это характерно для фторо-пласта-4 (см. рис. 2.22). Значения а-10*, °С\ при 9 = = 20...50°С и р = 0,1 МПа: 1) при 9 > 9с для эластомеров на базе НК 5,3 - 6,6; хлоропрена 4-5,5; СКН 3,3-5,3; СКЭП 4-6; СКТ 5,3-9,3; СКФ 5,8; 2) при 9 < 9с для эластомеров на базе НК 1,5; хлоропрена 1,65; СКЭП 1,2; СКФ 2; 3) для эбонита 0,7; капрона 1-1,5; текстолита 0,3; фторопласта-4 при 20 °С 2,5; при 60-120°С 1,2. Сжимаемость р = 1/х. В пределах одного физического состояния изменение относительного объема большинства полимеров происходит монотонно (рис. 2.16) и зависимость являет- ся экспоненциальной. Ее можно описать рядом линейных функций с модулями = + wiiP, в которых коэффициенты »1; = const в диапазонах р: 0 - 500, 500-1000, 1000 - 2000 МПа. При р > 2000 МПа X = х„ах = const. На кри- вых -у\р) для фторопласта существует излом при р « 800 МПа, отражающий полиформные превращения в материале. При повышении температуры К/ увеличивается, поэтому в пределах одного физического состояния следует ожидать уменьшения х с повышением 9. На рис. 2.17, а показаны типичные зависимости X (9) при р = const для термопластов (полистирол). Кривые имеют ступень при 9 = 9с, что отражает переход от аморфного состояния к высокоэласти- Х,ГЛа к, Г Па
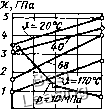 а, Г Па. 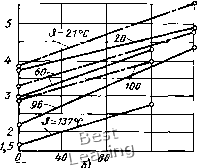 Таблица 2.2 50 100 ЦОр,ГПа О 50 100 150 С
о 40 80 120 160р, МПа 10 в В 120 р, МПа
200 Ш 600 40 , 60 9) 800 р, МПа -I-i 80 -J,"С Рис. 2.17. Зависимости модуля объемного сжатия полимеров от температуры Э (штриховые линии) и давления р (сплошные линии): а - для полистирола; б - для атактического полистирола и ПММА (штрихпунктирные линии); в - для ПЭНП; г - для ПЭВП; д - для синтетических каучуков (СКН, СКИ, СКС - заштрихованная зона) и вулканизованного НК [3] ческому И сопровождается ступенчатым уменьшением х. На рис. 2.17,6 показаны типичные зависимости х(р) при & = const для полимеров - ПММК и атактического полистирола в аморфном состоянии; на рис. 2.17, в и г - зависимости х(р) и х(Э) для частично-кристаллических термопластов - поли-этиленов ПЭВД и ПЭНД; на рис. 2.17, д-для эластомеров. Эти зависимости полностью или на определенных участках имеют линейный характер и могут быть описаны уравнениями х = Xq -I- тр, х = = Хо - фАО, хотя коэффициенты ш и ф существенно различны для разных материалов и интервалов температур. Конкретные данные существуют только по отдельным полимерам.- Учитывая известную аналогию в поведении полимеров и жидкостей для ориентировочных расчетов х в пределах одного физи- ческого состояния, можно использовать объединенное уравнение (1.5) в виде X = Хо -I- тр - ф АЭ, (2.8) где Хо - исходный модуль объемного сжатия при Ро = 0,1 МПа и Эо « 20 °С, Па; АЭ = Э - Эо, °С; m - безразмерный коэффициент; ф - коэффициент, зависящий от р и Э, Па/°С. Коэффициент т в зависимости от материала и температуры находится в пределах 8-15 (обычно m = 9...10) и незначительно меняется при изменении Э. Так, для эластомеров т к 10, для ПС 8,7, для ПММА 12, для ПЭВД 10, для ПЭНД 15. Следует иметь в виду, что данных по коэффициентам тиф опубликовано очень мало. Коэффициент ф, как правило, больше в области Э = 20...50°С (ф1 = 35... 15), меньше в области Э = 60...100°С (фг = Теплофизотедме свойства эластомеров [9]
Примечания. 1. Значения а/ приведены при Э > З;, (числитель) и 9<Эс (знаменатель). 2. Значения X приведены для наполненного вулканизата (резины). = 20...4) И слабо уменьшается с повышением р. Так, для эластомеров на основе НК ф1 = 33...27 и ф2 = 8; для ПС соответственно 22... 15 и 20... 12; для ПЭВД 20 и 4 для ПЭНД 22,5 и 25; для ПММА 17 и 8. Пример. Известны jco = 3500 МПа и «о = = 510"*°С" эластомера при р = 0,1 МПа и 9 = 20 °С. Определить х и а при р = = 50 МПа и 9 = 100 °С. Расчет выполняем по формулам (2.8), где т=10; ф1 = 30 (а = 20...50°С); (рг = 8 (9 = 50... 100°С) и (2.7): x = хо -I- тр - cpiASi - Ф2Д92 = = 3500 -ЫО 50 - 30 • 30 - 8 50 = 2700 МПа. а = аоио/и = 5 • Ю-*3500/2700 = = 6,5- ЮХ-. Эффект Томпсона. При быстром нагру-жении полимерный материал адиабатически нагревается. Мгновенное повышение температуры определяют по формуле АЭ = а (273 + 9) pi/c„, где с„ - тепло- емкость. При приложении давления р = 100 МПа образец нагревается на 15 -20°С. Через некоторое время (10- 15 мин) при р = const температура образца понижается до исходной. Удельная теплоемкость с = {dQ/dS-)/m, где Q - подведенное к телу количество теплоты; т - масса тела. Теплоемкость полимеров имеет ряд особенностей по сравнению с теплоемкостью обычньгх твердых тел, поскольку полимеры в зависимости от Э и р могут переходить из одного физического состояния в другое. Соответственно в структуре полимера может меняться число внутренних степеней свободы и удельный объем у, обусловливающие значения с. Для металлов с = Ср = с„ = = 0,13...0,9 кДж/(кг°С); для жидкостей с= 1,7...4,19 кДж/(кг-°С). Теплоемкость полимеров (табл. 2.2) в высокоэластичном и вязкотекучем состоянии близка к теплоемкости жид- Ср,нДж1(кг-к) 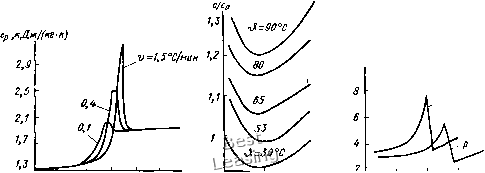 0,1 МПа = J4 10 го 30 4€ 50 60-Л°С zoo т р,МПа 50 100 150 200 J,°C В) в) Рис. 2.18. Зависимость теплоемкости полимеров с от температуры и давления р: а - для винилацетата; б - для ПММА; в - для ПЭВП [3] костей, в кристаллическом - к теплоемкости твердых тел, в стеклообразном - принимает промежуточные значения. При температурах кристаллизации и плавления с изменяется скачкообразно, в процессе стеклования - постепенно, но часто при &с кривые с(&) имеют максимум (с„ах), значение и положение которого зависят от скорости нагревания (рис. 2.18, а), в чем проявляется релаксационный характер процессов в полимерах. Теплоемкость полимеров существенно зависит от /7 и 9. На рис. 2.18, а показана типичная зависимость с (9) при р = const для области перехода из аморфного состояния в высокоэластическое. Для всех областей кроме области фазового перехода характерна линейная зависимость с = а + bp. На рис. 2.18, б и в показана зависимость с (9) для частично кристаллических полимеров: при 9 < 9ш, Ср увеличивается с повыщением 9, при 9 = 9пл Ср сначала резко уменьшается, затем постепенно увеличивается. При повышении р примерно до 200 МПа с сначала уменьшается, а затем незначительно увеличивается. Теплопроводность X, ВтДм • °С), материала обусловливает передаваемый через него тепловой поток Q, Вт: Q/t - - ХА TS/1, где S - площадь, м, / - толщина образца, м. Теплопроводность по- лимеров X, = 0,1...0,25 ВтДм-°С) (редко достигает 0,5): для ненаполненных резин 0,13-0,20; для ПТФЭ 0,2-0,25; для ПС 0,16-0,13, для ПЭНД 0,25-0,49). По виду температурной зависимости Х(9) полимеры можно разделить на несколько групп. Характерная для многих аморфных полимеров и НК зависимость показана на рис. 2.19, а: при 9 < 9с X увеличивается с увеличением 9, при 9 = 9с достигает максимума и при 9 > 9с уменьшается. Пределы изменения X с повышением р относительно невелики. Для полимеров в высокоэластическом состоянии характерна линейная зависимость [3]: X, = Х,о -I- Bp. Конкретных данных по зависимостям Х(9) и Х{р) для уплотнительных материалов недостаточно для обобщений. Теплофизические характеристики а, с, X смесей с наполнителями (Кн, с„, Xg) из- 0,8 0,6 «л 0,8 1,1 1,е в)
so S) р,НПа Рис. 2.19. Зависимость теплопроводности от температуры и давления для полимеров: а - СКТ, НК, ПММА, ПП и др.; б - ПС и ПТФЭ меняются по сравнению с характеристиками чистых эластомеров (а,, Сэ, Х,) в первом приближении пропорционально объемной доле w наполнителя [9]: а = аэ - W (аэ - а„); с = Сэ -I- w (сн - c,); X = Хэ -I- yw. 2.2. Резины и каучуки Эластомеры, получаемые на основе каучуков, называют резинами. В результате вулканизации резиновой смеси термопластичный, липкий и малопрочный каучук превращается в высокоэластичную прочную и стойкую во многих средах резину. Резина - термореактивный, пространственно сшитый сетчатый полимер с поперечными химическими связями между макромолекулами каучука. Комплекс механических и химических свойств резин уникален, поэтому они являются незаменимым материалом подавляющего большинства уплотнений и многих технических деталей. Природа механических свойств резин объясняется строением молекул каучука и характером химических и физических межмолекулярных связей. Основа резины - каучук - пластичное вещество (пластичностью называют свойство материала необратимо деформироваться под действием нагрузки). В невулкани-зованную (сырую) резиновую смесь путем механического смещения вводят ингредиенты: наполнители, вулканизующие агенты и др. При нагреве сырой резиновой смеси (вулканизации) между макромолекулами каучука возникают поперечные химические связи через атомы или группы вулканизующего агента (см. рис. 2.1, в). При вулканизации в пресс-форме деталь принимает форму ее рабочей полости. Макромолекулы каучука содержат порядка 10 звеньев, но лишь незначительная их часть оказывается «сшитой» между собою. В типичной резине одна поперечная связь приходится примерно на несколько сот звеньев цепи. Участки цепи между поперечными связями сохраняют гибкость и способность к движению с образованием многочисленных конформаций. При увеличении количества вулканизатора в смеси число связей возрастает, а резина становится менее эластичной вплоть до превращения в твердый и хрупкий эбонит. Пространственная сетка резины нерегулярная, поэтому при деформации возникают перенапряжения отдельных участков. Возникающие в них разрывы связей приводят к появлению первичных очагов разрушения, разрастающихся далее в трещины. Для предотвращения этого опасного явления в резиновую смесь вводят активные наполнители (часто называемые усилителями), которые представляют собою твердые мелкодисперсные вещества с большой площадью поверхности и поверхностной активностью (чаще всего технический углерод - сажу). Такие резины называют наполненными. В них между цепными макромолекулами кроме химических возникают адсорбционного характера связи с наполнителем, которые компенсируют нерегулярность поперечных химических связей. В перенапряженных участках пространственной структуры происходит сначала разрыв адсорбционных связей, которые затем восстанавливаются без разрушения материала, участок цепи разгружается, а не разрывается. Усилители значительно повышают прочность при растяжении, твердость, сопротивление истиранию и раз-диру резин на основе некристаллизую-щихся каучуков. Введение активных наполнителей в резины на основе кристаллизующихся каучуков существенно не влияет на прочность. При введении в резиновую смесь наполнителей уменьшается относительное содержание каучука в ней, т. е. снижается его расход и стоимость материала. Так как набухание резины в жидкостях определяется в основном свойствами каучука, смесь с большим содержанием наполнителей меньше набухает в рабочих средах. С этой целью в резиновые смеси кроме активных наполнителей 0 1 2 3 4 5 6 7 8 9 10 [ 11 ] 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||